Corallopyronin A and Myxopyronin B
Angew. Chem. Int. Ed.
2012, 51, 11381
A. Rentsch and M. Kalesse*
The retrosynthesis of corallopyronin begins with removal of
protecting groups (OTBS, and OCH2OTBS) and oxidation of the alcohol in 19 to the corresponding ketone. The OCH2OTBS group is called
tert-butyldimethylsiloxylmethyl group or SOM and was used for differentiating
two hydroxyl groups earlier in the synthesis.
Secondary alcohol 19 is
prepared by connecting fragments “A” and “B” by an alkylation reaction that
used LiTMP as the base.
Fragment “A” has a terminal conjugated aldehyde on the
right-hand-side that is installed by reduction of the corresponding ester to
the alcohol followed by oxidation to the aldehyde. The left-hand-side of fragment “A” has a
terminal propene that is formed by reacting acetaldehyde with the sulfone
formed by the oxidation of intermediate 18. Compound 18
is prepared by a Mitsunobu reaction between phenyltetrazole thiol 17 and terminal alcohol 16.
The TBS-protected secondary chiral alcohol of 16 is formed by using (-)-DIPCl as the chiral reduction agent upon
ketone 15. This reduction gave the product with 95% ee
and the absolute stereochemistry was confirmed by making the corresponding
Mosher’s esters. Ketone 15 is formed by reacting zincate 12 with aldehyde 14 followed by Swern oxidation of the resulting alcohol. Aldehyde 14
comes by a selective oxidative cleavage of the more electron-rich alkene in 13.
Finally, compound 13 is
prepared from Gerinol by an oxidation and Wittig protocol. The zincate intermediate 12 was prepared in
an interesting fashion from bromo-boronate 11
by treatment with dimethylzinc. In turn,
compound 11 is prepared from
bromo-alkyne 10, which I presume
comes from butynol.
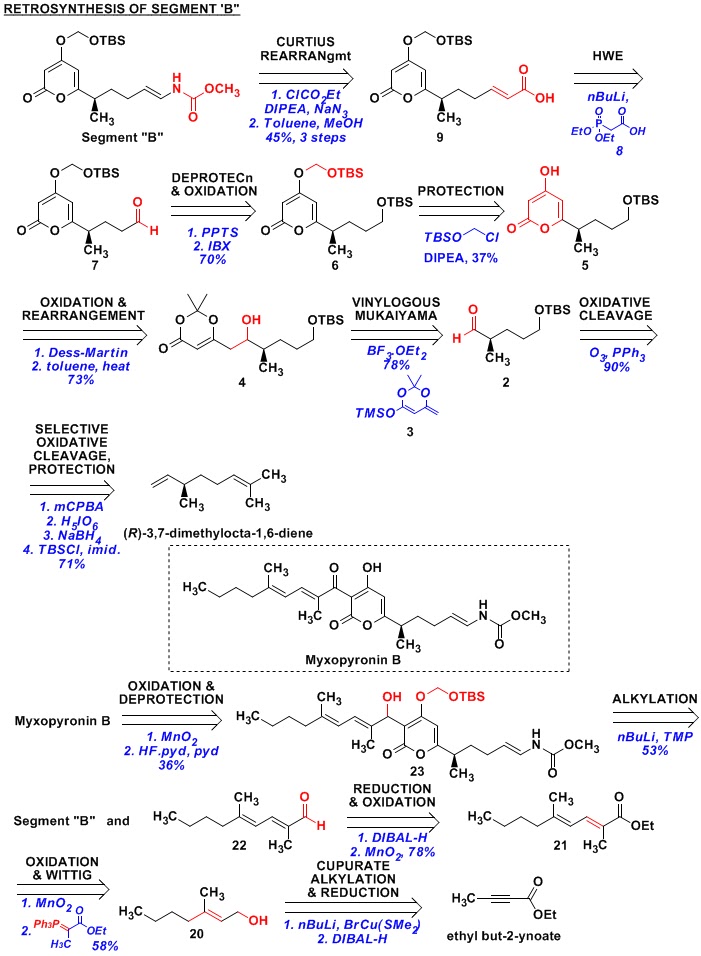
Fragment “B” has a terminal vinyl carbamate group on the
right-hand-side. Since vinyl-amines are
unstable, a Curtius-rearragement is used to prepare this functionality. This gives us acid 9 as the precursor, which is prepared from aldehyde 8 by a Horner-Wadsworth-Emmons reaction
of phosphonate ester 8 with aldehyde
7.
Aldehyde 7 is prepared from
TBS protected alcohol 6, which came from
protected alcohol 5. Here, we see the installation of the SOM
group to differentiate the terminal (TBS protected) alcohol from the hydroxyl
group on the enol-form of the diketoester.
Pyrone 5 comes from the
rearrangement of ketone formed by the oxidation of 4. Compound 4 is prepared by the signature reaction
of the Kalesse group – “the Vinylogous Mukaiyama Aldol Reaction” (VMAR). Thus, the reaction of aldehyde 3 with the
vinylogous-silyl-ketene-acetal 3
generates the desired product 4. Compound 2 is prepared from beta-(-)-citronellene by (a) selective
oxidative-cleavage of (R)-3,7-dimethylocta-1,6-diene; (b) reduction of the
aldehyde to the alcohol; and (c) protection of the alcohol as TBS ether.
In this paper, the authors have also described the
completion of the synthesis of a related natural product – Myxopyronin B. Retrosynthetically, it involves the
combination of Fragment “B” with aldehyde 22
(followed by oxidation and the removal of SOM protecting group). Aldehyde 22
is derived from ester 21, which in
turn, comes by an oxidation-Wittig sequence on alcohol 20. Alcohol 20 is prepared by the attack of nBu-cupurate (prepared by reacting nBuLi with BrCu(SMe2)) on
ethyl butynoate – an example of nBuLi
being used as the reagent with the “nBu” portion getting installed on the
molecule.
Overall, an excellent piece of work from the Kalesse group
which includes the signature “VMAR” (i.e. Vinylogous Mukaiyama Aldol
Reaction). Other interesting features
include the Curtius reaction which installs the sensitive vinylcarbamate group,
SOM groups that allows for the differentiation of hydroxyl groups, selective
oxidative cleavage of electron-rich alkenes in presence of electron-deficient
alkenes, liberal use of easy-to-perform oxidations and reductions, use of
(-)-DIPCl to establish the desired “R” configuration, using easily available chiral starting materials "geraniol" and "beta-(-)-citronellene", and finally, the use of
buffered HF.pyridine in THF/pyridine
to remove the silyl protecting groups.
The fact that all this was done by a single chemist makes it even more
commendable!